|
|
You are here: Foswiki>ABI Web>LectureWiki>PMSB_Seqan_2013>PSMB_Seqan_2013_Lagan (25 Apr 2013, RmaerkerUserTopic)Edit Attach
Page PSMB_Seqan_2013_Lagan
Hintergrund: Alignieren Genomischer Sequenzen
Durch das vergleichen zweier Genome unterschiedlicher Organismen können neue Erkenntnisse bezüglich der Konservierung funktionaler Einheiten gewonnen werden. Diese Erkenntnisse helfen uns Ursachen genetischer Krankheiten besser zu analysieren und zu verstehen. Um zwei biologische Sequenzen zu vergleichen wird ein globales Alignment berechnet. Dabei wird ermittelt welche Transformationen benötigt werden um eine Sequenz in die Andere zu überführen. Jedoch ist das standard Global-Alignment-Verfahren [1] nicht geeignet um genomische Sequenzen zu alignieren, da sich seine Laufzeit proportional zum Produkt der beiden Sequenzlängen verhält. Alternativ können lokale Alignments [2, 3] berechnet werden um homologe Regionen zu identifizieren. Es lassen sich so jedoch kaum Zusammenhänge zwischen der gemeinsamen Ordnung der funktionalen Einheiten ableiten. Um effizient genomische Sequenzen global zu alignieren, werden Heuristiken benutzt, die zu erst gute lokale Segmente zwischen den beiden Sequenzen filtern und anschließend diese Informationen nutzen um daraus ein globales Alignment zu berechnen. Einer der bekanntesten Vertreter dieser Heuristiken is der LAGAN-Algorithmus [4]. Dieser besteht im wesentlichen aus 3 Schirtten: 1) Generieren von lokalen Alignments zwischen den beiden Genomen. 2) Konstruieren einer globalen Map durch Chaining der identifizierten Segmente aus 1). 3) Berechnen des optimalen Alignments innerhalb der Region definiert durch die globale Map aus 2).Aufgaben
Ziel dieser Aufgabe ist es, den LAGAN-Algorithmus nach zu implementieren. Eingabe des Programms sind zwei FASTA-Dateien mit den beiden Genomen. Als Ausgabe soll das Programm das Alignment in einer Datei herausschreiben. Die entwickelten Methoden sollen sinnvoll getestet werden. Das Programm soll anhand eines Testdatensatzes auf Richtigkeit geprüft werden. Dafür werden zwei komplett distinkte Sequenzen generiert und anschließend zufällig erzeugte Seeds mit variabler Länge generiert. Jeder Seed wird zufällig in beide Sequenzen eingefügt und das Seed-Sequenz-Paar abgespeichert. Hinterher wird überprüft, ob korrekte Seed-Sequenz-Paare gefunden wurden. Im Anschluss soll der Algorithmus auf Realdaten angewendet werden. Je nach Fortschritt des Algorithmus sollen Sequenzen von verschiedenen Organismen aligniert werden:- 2 Phagen Genome (~
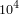 bp)
bp)
- 2 Bakterien Genome (~
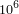 bp)
bp)
- 2 Hefen Genome (~
 bp)
bp)
- 2 Drosophila Genome (~
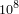 bp)
bp)
- Der 1. Teilnehmer berechnet alle lokalen Segmente zwischen den beiden Genomen mit dem STELLAR-Algorithmus [5].
- Der 2. Teilnehmer berechnet die globale Map mit dem Chaining-Algorithmus [6] und berechnet anschließend das Alignment mit Hilfe des BandedChain-Alignments [4].
- Der 3. Teilnehmer berechnet ebenfalls lokale Segmente jedoch mittel CHAOS-Chaining [7].
- Das Seeds2 Modul: Seed and Extend.
- Das I/O-Modul: Sequence I/O.
- Der STELLAR-Algorithmus: STELLAR.
References
- [1] http://en.wikipedia.org/wiki/Needleman%E2%80%93Wunsch_algorithm Needleman-Wunsch-Algorithmus
- [2] http://en.wikipedia.org/wiki/Smith-Waterman Smith-Waterman-Algorithmus
- [3] Basic Local Alignment Search Tool. Altschul, S.F., Gish, W., Miller, W., Myers, E.W., Lipman, D.J.(1990). J. Mol. Biol. 215:403–410.
- [4] LAGAN and Multi-LAGAN: Efficient Tools for Large-Scale Multiple Alignment of Genomic DNA. Brudno, M., et al. (2003). Genome Res. 2003. 13: 721-731. doi:10.1101/gr.926603
- [5] STELLAR: fast and exact local alignments. Kehr, B., Weese, D., Reinert, K. (2011). BMC Bioinformatics, 12(Suppl 9):S15, 2011.
- [6] Algorithms on Strings, Trees, and Sequences: Computer Science and Computational Biology. Gusfield, D., Cambridge, UK: Cambridge University Press; 1997
- [7] Fast and sensitive alignment of large genomic sequences. Brudno, M., Morgenstern, B., (2002). Bioinformatics Conference, 2002. Proceedings. IEEE Computer Society
Projektplanung und Umsetzung
ProjekthomepageComments
Edit | Attach | Print version | History: r5 < r4 < r3 < r2 | Backlinks | View wiki text | Edit wiki text | More topic actions
Topic revision: r4 - 25 Apr 2013, RmaerkerUserTopic
Ideas, requests, problems regarding Foswiki? Send feedback