|
|
You are here: Foswiki>ABI Web>LectureWiki>PMSB_Seqan_2014>PSMB_Seqan_2014_small_indels (16 Apr 2014, jsinger)Edit Attach
PSMB_Seqan_2014_small_indels
Hintergrund
 Neben SNPs sind kleine Insertionen und Deletionen (indels) eine Klasse wichtiger genomischer Varianten.
Für die Suche solcher Varianten bietet sich das split read mapping an.
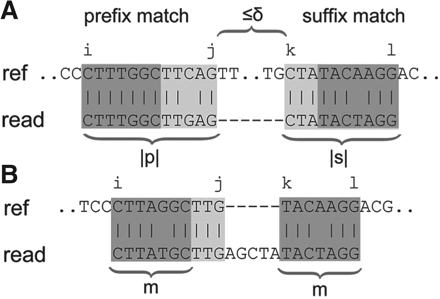
Dabei wird nicht ein ganzer Read mit möglichst kleinem Abstand in der Referenzsequenz gesucht sondern nur ein relativ kleines Präfix und Suffix (z.B. jeweils 16 Basen Präfix und Suffix von einem 100 bp Read) in der Referenz.
Wenn Präfix und Suffix bestimmte Abstandsbedingungen erfüllen, können über sog. split alignment die optimalen Breakpoints im Genom und Read bestimmt werden.
So ergibt sich auch die genaue Position und Sequenz der Insertion bzw. Deletion.
Neben SNPs sind kleine Insertionen und Deletionen (indels) eine Klasse wichtiger genomischer Varianten.
Für die Suche solcher Varianten bietet sich das split read mapping an.
Dabei wird nicht ein ganzer Read mit möglichst kleinem Abstand in der Referenzsequenz gesucht sondern nur ein relativ kleines Präfix und Suffix (z.B. jeweils 16 Basen Präfix und Suffix von einem 100 bp Read) in der Referenz.
Wenn Präfix und Suffix bestimmte Abstandsbedingungen erfüllen, können über sog. split alignment die optimalen Breakpoints im Genom und Read bestimmt werden.
So ergibt sich auch die genaue Position und Sequenz der Insertion bzw. Deletion.
Aufgaben
Es soll ein einfacher Split Read Mapper implementiert werden. Eingabe ist die Referenz sowie die Reads, annotiert mit ihrem ursprünglichen Alignment aus dem ersten Read Mapping Schritt bzw. der Information, dass sie nicht aliniert werden konnten. Es ist dann ein Prefix bzw. Suffix exakt oder approximativ zu suchen. Für Paare von Prefix/Suffixmatches mit passendem Abstand soll dann split alignment ausgeführt werden um die Breakpoints und Sequenz zu bestimmen. Als Ausgabe sollen die Matches aus der Eingabe um die neu gefundenen split matches erweitert werden. Über das Clustern von split matches können die kleinen indels genauer bestimmt werden. Die Qualität und Performance kann mit existierenden Tools wie SplazerS verglichen werden. Das Modul soll sowohl aus Dateien lesen bzw. in Dateien schreiben können wie sich auch direkt in das globale Pipelineprogramm integrieren.References
- Anne-Katrin Emde, David Weese, Marcel Schulz, Stefan Haas, and Knut Reinert (2012). Detecting genomic indel variants with exact breakpoints in single- and paired-end sequencing data using SplazerS. Bioinformatics.
Mindestanforderungen:
- Einlesen der Referenz, der Reads und der Mapping Informationen
- Finden exakter Pre-/Suffixe der Reads
- Berechnung valider Paare (ueber den Abstand)
- Mit Hilfe von "splitAligment" das Split-Alignment berechnen
- Breakpoint bestimmen
- Speichern der Ergebnisse
- Testen
- Dokumentieren
Zusatz:
- Vergleicht mit bestehenden tools
- Finden approximativer Pre-/Suffixe
- Statistik ueber Insertlaengen
- Die funktion "splitAlignment" durch eigene Implementierung ersetzen
Expose
Fortschrittsbericht
- Split_Read_Mapping.gif:


| I | Attachment | Action | Size | Date | Who | Comment |
|---|---|---|---|---|---|---|
| |
Split_Read_Mapping.gif | manage | 34 K | 01 Apr 2014 - 11:09 | UnknownUser |
{kind=link}
Edit | Attach | Print version | History: r6 < r5 < r4 < r3 | Backlinks | View wiki text | Edit wiki text | More topic actions
Topic revision: r5 - 16 Apr 2014, jsinger
Ideas, requests, problems regarding Foswiki? Send feedback