|
|
You are here: Foswiki>ABI Web>ThesesHome>BscImprovementsOfGraphBasedRealignment (20 Jun 2013, holtgrew)Edit Attach
BscImprovementsOfGraphBasedRealignment
Improving the Graph-Based Realignment in SeqAn.Background
The SeqAn library contains a powerful method for realignment and consensus based on the alignment graph [1]. While this works quite well, the approach has some drawbacks. The following picture shows a multi-read alignment where the graph-based (re-)alignment method did not succeed in create a good alignment.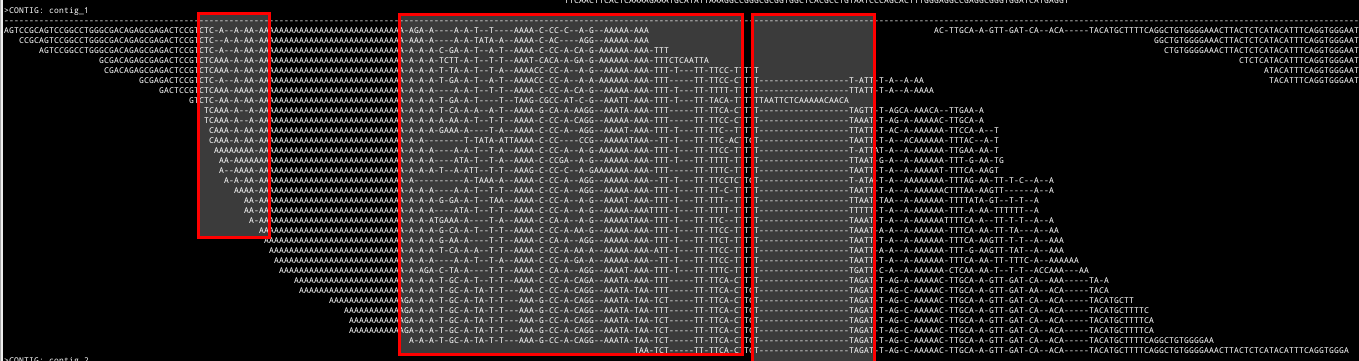 The input for the graph-based alignment are matches between the reads.
The matches might be conflicting and the alignment algorithm selects some matches while discarding others.
In the left two marked regions, this leads to many small insertions and deletions while in the right marked region, there is a long stretch
The input for the graph-based alignment are matches between the reads.
The matches might be conflicting and the alignment algorithm selects some matches while discarding others.
In the left two marked regions, this leads to many small insertions and deletions while in the right marked region, there is a long stretch TAATT...CAACA that the matches were discarded for.
Another problem of the realignment method is the cubic running time of the triplet consensus extension.
This is problematic with deep alignments (hundreds of stacked sequences).
Topic
The aim of this thesis is to fix the issues mentioned above.- The artifacts could be fixed in a postprocessing step. The student should develop heuristics to find such artifacts and fix them (e.g. creating pairwise read alignments in the problematic regions, add them to the underlying alignment graph, and perform progressive alignemtn again).
- The running time problems could be accommodated by doing the consensus extension in a hierarchical fashion or with sampling.
Comments
References
- [1] Rausch, Tobias, et al. A consistency-based consensus algorithm for de novo and reference-guided sequence assembly of short reads. Bioinformatics 25.9 (2009): 1118-1124.


| I | Attachment | Action | Size | Date | Who | Comment |
|---|---|---|---|---|---|---|
| |
multi_read_alignment.png | manage | 55 K | 20 Jun 2013 - 10:16 | UnknownUser |
{kind=link}
Edit | Attach | Print version | History: r4 < r3 < r2 < r1 | Backlinks | View wiki text | Edit wiki text | More topic actions
Topic revision: r3 - 20 Jun 2013, holtgrew
Ideas, requests, problems regarding Foswiki? Send feedback