
Theoretical and Mathematical Physics in Molecular Simulation
Office
Dorothé Auth
Project leader: Prof. Dr. Luigi Delle Site
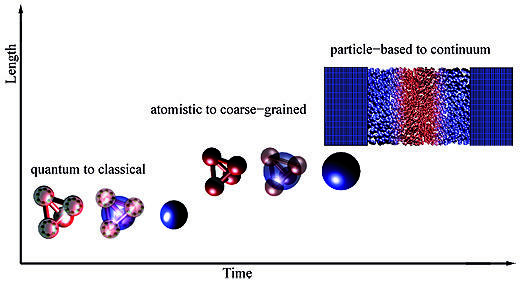
The focus of the group is the development of computational methodologies for molecular simulations which link different scales in problems occurring in condensed matter, material science and chemical physics.
Such theoretical/computational tools are able to identify and treat only those details that are strictly required by the problem and thus can describe the interplay of scales in a satisfactory way.
Our current research aims to link in a concurrent way quantum, atomistic, coarse-grained and continuum resolution for investigating properties of complex molecular systems over a large set of thermodynamic conditions.
For more details, please visit the link.