|
|
You are here: Foswiki>ABI Web>VariantDetection>VariantDetectionPapers (18 May 2011, AnneKatrinEmde)Edit Attach
Page VariantDetectionPapers
Remarks to papers concerning variant detection in NGS data.- Method Papers
- Simple variation
- Pairede-end mapping based
- Combinatorial algorithms for structural variation detection in high-throughput sequenced genomes
- BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nature Methods 2009.
- PEMer: a computational framework with simulation-based error models for inferring genomic structural variants from massive paired-end sequencing data. Genome Biology 2009.
- SVDetect: a tool to identify genomic structural variations from paired-end and mate-pair sequencing data. Bioinformatics 2010.
- MoDIL: detecting small indels from clone-end sequencing with mixtures of distributions. Nature Methods 2009.
- Depth-of-coverage (DOC) methods
- Split/Spliced-read mapping
- Fast and SNP-tolerant detection of complex variants and splicing in short reads
- SuperSplat
- Global und unbiased detection of splice junctions from RNA-seq data
- Detection of splice junctions from paired-end RNA-seq data by SpliceMap
- MapSplice: Accurate mapping of RNA-seq reads for splice junction discovery
- A robust framework for detecting structural variations in a genome
- Comments
Method Papers
Simple variation
VarScan: variant detection in massively parallel sequencing of individual and pooled samples
Daniel C. Koboldt∗, Ken Chen, Todd Wylie, David E. Larson, Michael D. McLellan, Elaine R. Mardis, George M. Weinstock, Richard K. Wilson and Li Ding Tool Name: VarScan (available for download) Variation Types: SNPs, indels Assumptions: Illumina/454 Method: "Given an alignments file, VarScan scores and sorts the alignments on a perread basis, discarding reads that aligned with low identity or to multiple (ambiguous) locations in the reference sequence. Next, the single best alignment for each read is screened for sequence changes. Variants detected in multiple reads are then combined together into unique SNPs and indels. For each predicted variant, VarScan determines the overall coverage, as well as the number of supporting reads, average base quality, and number of strands observed for each allele. Thresholds for coverage, quality, variant frequency, and/or number of reads required to call a variant are set automatically with the easyrun command, but can be manually adjusted by the user. VarScan reports SNPs, insertions, and deletions with their chromosomal coordinates, alleles, flanking sequence, and read counts." Comments: "Here, we describe VarScan, an open source tool for detecting SNPs, insertions, and deletions and assessing their frequencies in massively parallel sequencing data. Unlike currently available variant detection tools, VarScan is compatible with several read aligners (BLAT, Newbler, cross_match, Bowtie and Novoalign), and calls variants in both individual and pooled samples."SNP detection for massively parallel whole-genome resequencing
Ruiqiang Li, Yingrui Li, Xiaodong Fang, Huanming Yang, Jian Wang, Karsten Kristiansen and Jun Wang Tool Name: SOAPsnp (available for download) Variation Types: SNPs Assumptions: Illumina sequencing data Method:- Bayesian genotype calling (for each position likelihood is computed, priors are set according to the observation that transitions are four times more frequent than transversions)
- Recalibration of quality values according to observed mismatch rate
- Account for intrinsic bias of errors in Illumina data
- Sum rank test is used to eliminate artificial heterozygote calls
Bayesian model:
- posterior probability of genotype
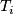 :
: 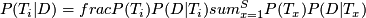
- likelihood of diploid genotype
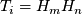 :
: 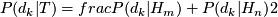
- likelihood of
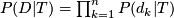
- Low false positive rate for any sequencing depth
galign: A tool for rapid genome polymorphism discovery
Shai Shaham September 2009 PLoS ONEDindel: Accurate indel calls from short-read data
Cornelis A. Albers, Gerton Lunter, Daniel G. MacArthur, Gliean McVean, Willem H. Ouwehand, and Richard Durbin. Genome Reserach Oct 2010 Tool Name: Dindel Variation Type: small indels, SNPs Assumptions: Illumina data Method:produces candidate haplotypes of (indel) candidate regions, realigns reads to haplotypes (iteratively, using EM algo), and assign quality scores to indel calls.
Input: requires mapped reads (BAM) with mapping qualities.Pairede-end mapping based
Combinatorial algorithms for structural variation detection in high-throughput sequenced genomes
Fereydoun Hormozdiari, Can Alkan, Evan E. Eichler, and S. Cenk Sahinalp Tool Name: ? Variation Type: indels, inversions (SVs) Assumptions: PE data Method: ?BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nature Methods 2009.
Ken Chen, John W Wallis, Michael D McLellan, … Elaine R Mardis. Tool Name: BreakDancerPEMer: a computational framework with simulation-based error models for inferring genomic structural variants from massive paired-end sequencing data. Genome Biology 2009.
Jan O Korbel, Alexej Abyzov, Xinmeng J Mu, … Michael Snyder and Mark B Gerstein. Tool Name: PEMerSVDetect: a tool to identify genomic structural variations from paired-end and mate-pair sequencing data. Bioinformatics 2010.
Bruno Zeitouni, Valentina Boeva, Isabelle Janoueix-Lerosey, … Emmanuel Barillot. Tool Name: SVDetectMoDIL: detecting small indels from clone-end sequencing with mixtures of distributions. Nature Methods 2009.
Seunghak Lee, Fereydoun Hormozdiari, Can Alkan and Michael Brudno. Tool Name: MoDILDepth-of-coverage (DOC) methods
Sensitive and accurate detection of copy number variants using read depth of coverage
Seungtai Yoon, Zhenyu Xuan, Vladimir Makarov, et al. August 2009 Genome Research Tool Name: EWT (not available for download) Variation Type: CNVs Assumptions: High coverage (30x) → normality in 100bp windows Method: EWT (event wise testing). Number of read starts (read depth RD) in non-overlapping 100bp windows.- For each window:
 is corrected according to GC content in window
is corrected according to GC content in window 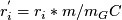 where
where 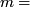 median RD,
median RD, 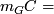 median RD for windows with this GC. Z-score:
median RD for windows with this GC. Z-score: 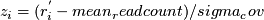 . Upper/lower tail probability:
. Upper/lower tail probability: 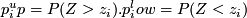 .
. - For an interval A of consecutive windows:
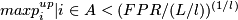 unusual event, deletion:
unusual event, deletion: 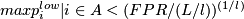 where FPR is the nominal false-positive rate,
where FPR is the nominal false-positive rate, 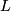 is the number of windows of a chromosome,
is the number of windows of a chromosome, 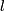 is the size of the interval A.
is the size of the interval A. - Iteration over l until N-1 where
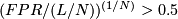
- Merging of events, cluster formation: events within 500bp proximity are merged (if copy number change in the same direction). events with low absolute difference from the average (compare median rd in event vs global average) are discarded (if within 0.75..1.25). Significance of each cluster is tested by a one-sided Z-test (significance level
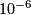 )
)
- Pairwise comparison of RD data from multiple individuals: RD in detected 100bp windows are compared by t-test, if p-values < 0.001 and absolute difference between read counts > 0.5 --> polymorphic.
- Copy number = round(2*r_i^'/mean_readcount)
Difference arrayCGH ↔ RD data: for array data variance in probe ratios is lowest for normal state, variance increases with copy number changes in both directions. for RD data, variance is lowest for the deletion state and increases proportionally with copy number.
Overlap PE detected SVs ↔ RD detected SVs: very low (5-10%)! PE SVs enriched in simple repeats (12% of predicted PE SVs), RD SVs enriched in segmental duplications (40% of predicted RD SVs)
Implementation: Java Comments:- inconsistencies: the authors first write that reads with mapping quality < 30 were filtered out (MAQ alignments). later they say that due to maq's random distribution of multi-reads coverage in repetitive regions is not different from the mean coverage. ???
Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs
Joshua M Korn, Finny G Kuruvilla, Steven A McCarroll, Alec Wysoker, James Nemesh, Simon Cawley, Earl Hubbell, Jim Veitch, Patrick J Collins, Katayoon Darvishi, Charles Lee, Marcia M Nizzari, Stacey B Gabriel, Shaun Purcell, Mark J Daly, and David Altshuler October 2008 Nature Genetics Tool Name: Birdsuite (available for download) Variation Types: SNPs, CNVs Assumptions: SNP array data, but ideas should be applicable to sequence data Method:- CNP genotyping: Assign copy number across regions of known CNVs
- SNP genotyping: Assign diploid genotypes to all regions having copy number two (+ detect probe-specific means and variances)
- CNV discovery: Rare CNV detection by using probe-sepcific means and variances
- Use CN information to estimate genotypes such as AAB and A
Split/Spliced-read mapping
Fast and SNP-tolerant detection of complex variants and splicing in short reads
Thomas D. Wu and Serban Nacu Februray 2010, Bioinformatics Tool Name: GSNAP Implementation: C and Perl Variation Types: complex variants, splicing Assumptions: Method:- SNP-tolerant: aligning to a reference "space" (major/minor-allele genome)
- index 12-mers every 3 nt in the genome (including all known SNP combinations)
- spanning/complete set method to filter candidate regions
- "full" sensitivity (as if every 12-mer were indexed) (100% sensitivity actually only guaranteed when read and ref share a common 14-mer --> floor(readLen/14)-1 mismatches)
- ignores 12-mers that occur more than 10 times the mean (greedy option for subsequently including them, if no alignment is found)
- indel can be close to the end of the read (vs our method: no shorter than q!)
- probabilistic model for splice sites. if aligned exon sequence on one side is short, splice score has to be higher. if aligned sequence is long, splice score can be lower
- also reports interchromosomal splice junctions and "partial-splicing"-matches
- PE mode
- 100K reads from whole genome, read lengths 36, 70, 100. ins up tp 9bp, del up to 30bp: compared with BWA, Bowtie, SOAP2, SOAP, MAQ. most sensitive for indels, runtimes comparable
- simluated spliced reads based on RefSeq splice sites: missed only 0.1% of intragenic and 5% of intergenic splice sites (due to ow splice scores)
- known-SNP-tolerance increases alignment yield by 0.5%, 5% of reads were found to have additional best mapping locations, 0.3% of reads could be disambiguified by SNP tolerance
SuperSplat
Tool Name: SuperSlat Implementation: c Variation Types: splice junctions Assumptions: Method: q-gram index of reference. No mismatches in alignment: "Supersplat works by partitioning each read into all possible two-chunk sizes, given minimum read chunk size (-c), and searching over each reference for locations where both chunks exactly match the reference, within given minimum intron size (-n) and maximum intron size (x)." Comments: output can become extensive! outputs every possible split position on a read, i.e. each read multiple timesGlobal und unbiased detection of splice junctions from RNA-seq data
Tool Name: SplitSeek Implementation: Perl Variation Types: splice junctions Assumptions: Method:- uses Applied Biosystems tools, splits reads into prefix-anchor, gap, and suffix-anchor. anchors are aligned separately, only reads where both anchors map uniquely are from then on considered!! combines anchor matches in perl script (extending half-matched reads into other exon if supported by other reads)
- can identify junctions reads that overlap >= 5 bp with exon
- low FDR (0.1%)
- compare with RNA-MATE ?
Detection of splice junctions from paired-end RNA-seq data by SpliceMap
Tool Name: SpliceMap Implementation: Python Variation Types: splice junctions Assumptions: Method:- Half-read mapping
- Seeding selection
- Junction search (* Paired-end filtering)
MapSplice: Accurate mapping of RNA-seq reads for splice junction discovery
Kai Wang, Darshan Singh, Zheng Zeng,Stephen J. Coleman, … , Jan F. Prins and Jinze Liu. June 2010 Nucleic Acids Research Tool Name: MapSplice Implementation: Python Variation Types: splice junctions Assumptions: Method:first part: single read mapping
- partition reads into segments
- (exonic?) alignment of segments (using Bowtie), keep multi-hits
- spliced alignment of segments, double-anchored (first and last segment of read match, but middle one doesnt) or single-anchored (only one neighboring segment matches, search within maxDist window)
- segment assembly, find best split position, assign quality value to read (simple mismatch counts basically, but somehow quality weighted)
second part: multi-read assignment
- splice junction inference: each read gets an anchor significance score, dependent on the length of the shortest segment. each candidate splice junction get a significance score: the max anchor score of supporting reads, a shannon entropy score somehow rewarding uniformity of mapped reads, and an average read quality, somehow combined.
- select best alignment for each read: combine read quality socre + junction quality score
A robust framework for detecting structural variations in a genome
Seunghak Lee, Elango Cheran and Michael Brudno 2008 BioinformaticsComments
Edit | Attach | Print version | History: r21 < r20 < r19 < r18 | Backlinks | View wiki text | Edit wiki text | More topic actions
Topic revision: r20 - 18 May 2011, AnneKatrinEmde
- This page was cached on 11 Mar 2025 - 08:36.
Ideas, requests, problems regarding Foswiki? Send feedback