|
|
You are here: Foswiki>ABI Web>LectureWiki>PMSB_Seqan_2015>PSMB_Seqan_2015_palign (12 Mar 2015, RmaerkerUserTopic)Edit Attach
PSMB_Seqan_2015_palign
Einleitung
 Paarweise Sequenzalignments liefern wertvolle biologische Informationen über die Identität zweier Sequenzen.
Entsprechend werden sie in vielen Programmen und Algorithmen verwendet.
Einer der bekanntesten Algorithmen zum lösen eines paarweisen Sequenzvergleiches ist der Needleman-Wunsch Algorithmus [1].
Jedoch benötigt dieser quadratische Laufzeit und quadratischen Speicherbedarf.
Paarweise Sequenzalignments liefern wertvolle biologische Informationen über die Identität zweier Sequenzen.
Entsprechend werden sie in vielen Programmen und Algorithmen verwendet.
Einer der bekanntesten Algorithmen zum lösen eines paarweisen Sequenzvergleiches ist der Needleman-Wunsch Algorithmus [1].
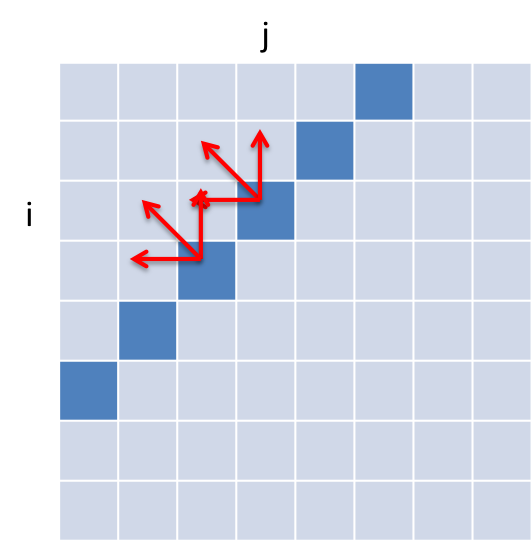
Jedoch benötigt dieser quadratische Laufzeit und quadratischen Speicherbedarf. Allerdings haben sich auch die Computer stark weiterentwickelt. Heutzutage besitzen normale PCs mehrere Rechenkerne die es ermöglichen Befehle parallel auszuführen. In diesem Projekt soll der Needleman-Wunsch Algorithmus mittels OpenMP parallelisiert werden. Der typische Ansatz um die DP-Matrix zu parallelisieren ist es die Antidiagonalen zu berechnen (siehe Bild), da hier zwischen den Zellen keine Datenabhängigkeiten existieren.
Aufgaben
Im Rahmen dieses Softwareprojektes soll der Needleman-Wunsch Algorithmus mit OpenMP parallelisiert werden. Es soll ein Programm geschrieben werden, das 2 Sequenzen einliest und zwischen diesen ein Alignment berechnet. Des Weiteren wird dem Programm die Werte für die Scoringmatrix sowie die Anzahl der Threads übergeben. Das Programm berechnet das Alignment und schreibt das Ergebnis (Score und Alignment) in eine Datei, die von dem Nutzer übergeben wurde. Anschließend soll die Laufzeit des parallelisierten Needleman-Wunsch mit dem seriellen aus der SeqAn-Bibliothek verglichen werden. Dabei sollen verschiedene Testdatensätze evaluiert und grafisch Dargestellt werden.Zusatz
Option A
Das Programm soll affine Gapkosten unterstützen.Option B
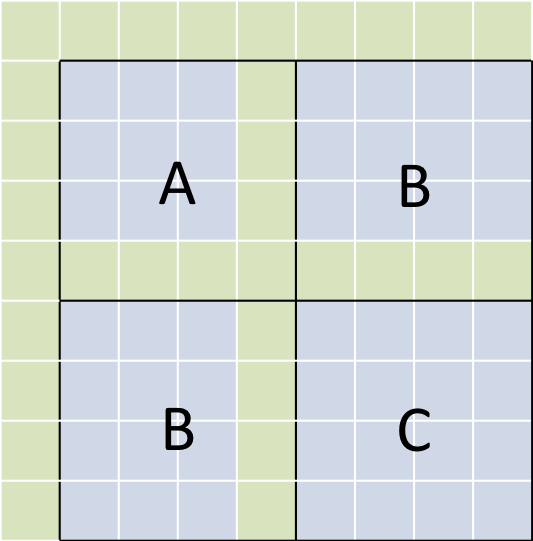 Eine weitere Möglichkeit die Parallelisierung zu verbessern ist das sogenannte tiling [2]. Dabei wird die Matrix rekursiv in kleinere Blöcke geteilt, sodass diese später Cache effizient berechnet werden können. Während jeder Block seriell berechnet wird, können antidiagonale Blöcke parallel prozessiert werden.
Eine weitere Möglichkeit die Parallelisierung zu verbessern ist das sogenannte tiling [2]. Dabei wird die Matrix rekursiv in kleinere Blöcke geteilt, sodass diese später Cache effizient berechnet werden können. Während jeder Block seriell berechnet wird, können antidiagonale Blöcke parallel prozessiert werden. In der Erweiterung soll dem Program ein weiterer Parameter für die Blockgröße übergeben werden. Die Matrix soll dann iterative in die Blöcke geteilt und anschließend die Blöcke in Antidiagonalen parallel berechnet werden.
Evaluation
Die entwickelten Methoden sollen sinnvoll getestet werden. Das Programm soll anhand eines Testdatensatzes auf Richtigkeit geprüft werden. Im Anschluss soll der Algorithmus auf Realdaten angewendet werden. Je nach Fortschritt des Algorithmus sollen Sequenzen von verschiedenen Organismen verglichen werden:- 2 Phagen Genome (~
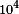 bp)
bp)
- 2 Bakterien Genome (~
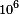 bp)
bp)
SeqAn Module
Die folgenden Teile von SeqAn sind relevant:- Das I/O-Modul: Sequence I/O.
- Das Alignment-Modul: Alignment Representations.
Referenzen
[1] Needleman, Saul B., and Christian D. Wunsch. "A general method applicable to the search for similarities in the amino acid sequence of two proteins." Journal of molecular biology 48.3 (1970): 443-453. [2] Chowdhury, Rezaul Alan, Hai-Son Le, and Vijaya Ramachandran. "Cache-oblivious dynamic programming for bioinformatics." IEEE/ACM Transactions on Computational Biology and Bioinformatics (TCBB) 7.3 (2010): 495-510.Edit | Attach | Print version | History: r2 < r1 | Backlinks | View wiki text | Edit wiki text | More topic actions
Topic revision: r1 - 12 Mar 2015, RmaerkerUserTopic
Ideas, requests, problems regarding Foswiki? Send feedback