Implicit solvation models in electronic structure simulations
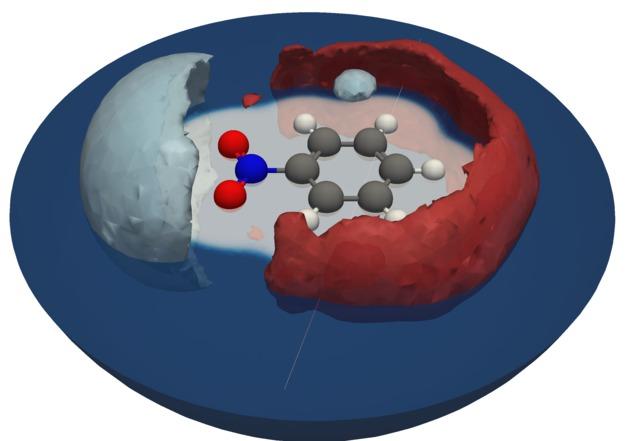
Electronic structure methods such as Density Functional Theory and beyond are the most accurate way for describing chemical bonding. By their attempt to approximate the electronic Schrödiger equation, these come with considerable computational costs. When targeting at situations, where a solvent is present, e. g. at a liquid-solid interface, these costs easily explode owing to the need to sufficiently sample the molecular motion within the solvent. One efficient way to circumvent this problem are implicit solvation models. Here the solvent is modeled as a continuum, and the corresponding (non-linear) partial differential equations are solved parallel to the governing equations of the electronic structure method. This removes the need for sampling but also many degrees of freedom. We improve existing solvation models and develop numerical schemes for the solution of the corresponding PDE. For the later, we employ overlapping atom-centered radial grids and a multi-center multipole expansion basis. By this choice, we can force the appropriate far-field asymptotics and can operate on the same computational structures as many full-electron codes. We thereby avoid mesh generation for the solvation model and the interpolation between different grids and arrive at an efficient formulation.
Publications
Sinstein, M., Scheurer, C., Matera, S., Blum, V., Reuter, K., & Oberhofer, H. (2017). Efficient Implicit Solvation Method for Full Potential DFT. Journal of chemical theory and computation, 13(11), 5582-5603.
Ringe, S., Oberhofer, H., Hille, C., Matera, S., & Reuter, K. (2016). Function-Space-Based Solution Scheme for the Size-Modified Poisson–Boltzmann Equation in Full-Potential DFT. Journal of chemical theory and computation, 12(8), 4052-4066